Brainstem Infarct Presenting As Torsades De Pointes
Sivakumar Ardhanari, BalashankarGomathi S, Anita L, Dr. Harish A,
Mullasari Ajit S, Ulhas M. Pandurangi
Institute of Cardiovascular Diseases,Madras Medical Mission, 4A, Dr. J.J.Nagar, Mogappair, Chennai - 600 037
|
|
INTRODUCTION
Central Neurogenic Hyperventilation (CNH) is an unusual presentation of acute brainstem infarct.1,2 The resultant respiratory alkalosis may cause significant hypokalemia which predisposes in susceptible individuals to prolongation of cardiac repolarisation phase and torsades de pointes (TdP).3,4 The hemodynamic instability due to recurrent TdP may result in hypoxic encephalopathy which makes the accurate localization of central nervous system lesion difficult. We report a case of recurrent TdP due primarily to brain stem infarct, the diagnosis of which was masked by features of hypoxic encephalopathy. The diagnosis of brainstem infarct could be confirmed only when neurological deficits persisted long after suppression of TdP and achievement of hemodynamic stability.
Case report:
A 38 years old lady with history of diabetes mellitus for 10 years was referred to us for management of refractory ventricular tachyarrhythmia. The medical report of the referring hospital indicated that the patient presented with history of sudden onset of altered sensorium of one-hour duration. She was reported to have recurrent polymorphic ventricular tachycardia despite several attempts of DC shocks, intravenous lignocaine and amiodarone. On transfer to our centre within two hours, she was found to be restless, incoherent and tachypneic. There were no obvious localizing neurological signs. The pulse and blood pressure were not recordable. Cardiac rhythm monitor showed recurrent polymorphic wide QRS tachycardia (Fig 1).
Sinus rhythm (Fig 2) was transiently achieved by repetitive DC shocks and QT interval was found to be prolonged (666 ms). In view of long QT interval and polymorphic, wide QRS morphology, the tachycardia was diagnosed as TdP.
Detailed history obtained from the attending family members suggested that the patient had developed sudden onset incoherence and inability to stand erect followed by laboured breathing. There was no history of fever, vomiting, diarrhea, headache, poisoning or trauma. Apart from her regular insulin there was no history of intake of any other medications. The arterial blood gas analysis showed respiratory alkalosis - pH of 7.63, PaO2 of 190.2 mm Hg, PaCO2, 9.3 mm Hg; and bicarbonate of 10.5 mEq/L. Serum electrolyte analysis showed low normal levels of magnesium (1.7 mEq/L), normal level of sodium (138 mEq/L) with significant hypokalemia (2.4 mEq/L). The blood glucose level, renal and liver function tests were within normal limits.
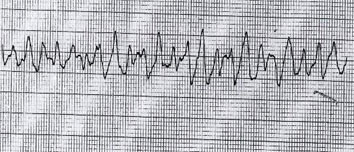 |
Fig 1. Cardiac rhythm strip on admission showing typical ‘twisting’ QRS morphology of TdP. |
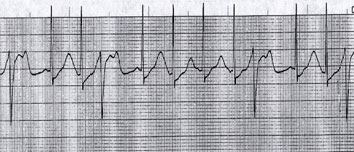 |
| Fig 2. Cardiac rhythm strip after cardioversion showing sinus rhythm with long QTc (666 ms). Note frequent ventricular ectopics during ‘vulnerable phase’ (R on T PVCs) which can trigger TdP. |
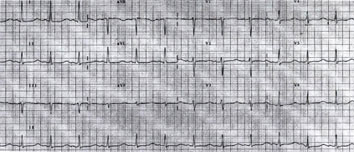 |
| Fig 3. Surface 12 lead ECG obtained 3 hours after admission showing persistence of QT prolongation (670 ms). |
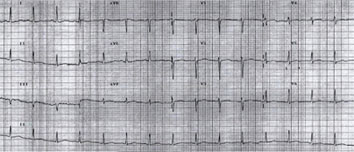 |
| Fig 4. Surface 12 lead ECG after 24 hours of admission and correction of electrolytes. Note normalization of QT interval (430ms). |
Correspondence: Dr.Yatin Mehta,
Sr. Consultant Anaesthesiologist
Indraprastha Apollo Hospitals, New Delhi
Email: yatinmehta@hotmail.com
|
Need for DC shock used to arise almost every five minutes. Intravenous correction of potassium levels was initiated. Prophylactic intravenous magnesium infusion was started to achieve magnesium level 2 mEq/L. Temporary pacemaker lead was inserted and attempts were made to keep the ventricular rate of 120 bpm. Over a period of three hours, almost 30 DC shocks were delivered before sinus rhythm (Fig 3) was achieved. However QT interval remained prolonged (QTc 670 ms). Over drive ventricular pacing at 100 bpm was continued to suppress ventricular ectopic activity.
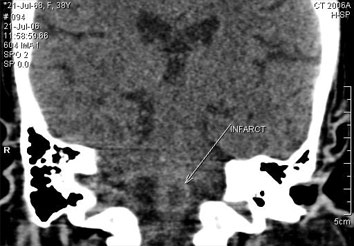
Patient remained incoherent, tachypneic with no definite localizing neurological signs. Respiratory alkalosis persisted. These abnormal neurological features were attributed to hypoxic encephalopathy. Even after achieving normal serum electrolyte levels, stable sinus rhythm and normalization of QT interval (Fig 4) with stable hemodynamics for 24 hours, she continued to be disoriented and tachypneic, raising suspicion of intracranial pathology. A CT scan was ordered, which revealed a hypodense area (20 HU) of size 6mm in the medulla oblongata suggestive of ischemic infarct (Fig 5). Her consciousness progressively deteriorated and she ultimately succumbed to respiratory arrest.
Discussion:
Torsades de pointes (TdP) is a polymorphic ventricular tachycardia (VT), which was described for the first time by Dessertenne in the year 1966. It has a characteristic QRS morphology, wherein QRS complexes (‘pointes’) twist around isoelectric line during the arrhythmia.5 It is associated with prolongation of ventricular repolarisation phase of action potential curve, represented by long QT interval on the ECG during sinus rhythm.6 The mechanisms underlying TdP are far from understood. It is believed that dysregulation of inward potassium current (IKr) by certain ion channels lead to prolongation of action potential duration (APD). These ion channels are more often encoded by HERG gene. Prolongation of APD (long QT interval) precipitates early after depolarization and triggered activity which initiate TdP. 7,8
 |
The syndrome of long QT interval (LQTS) and TdP can be inherited or acquired. 9 The inherited forms of LQTS can be autosomal dominant (Romano-Ward syndrome) or autosomal recessive (Jervell-Lange-Nielsen syndrome). The acquired forms of LQTS are largely drug induced. Antiarrhythmic drugs (Vaughan Williams Class I and III), tricyclic antidepressants, phenothiazines and macrolide antibiotics are the drugs which are implicated most often. Certain metabolic disturbances (hypokalemia, hypocalcemia and hypomagnesemia) also predispose to LQTS in susceptible individuals. The other common causes of acquired LQTS and TdP include cerebrovascular diseases like stroke even in the absence of metabolic disturbances, altered nutritional state and poisoning.4 Genetic analysis may reveal inherited forms of LQTS in some of the susceptible individuals.9
The clinical aspects of TdP are heterogeneous. Attacks of TdP may differ in their frequency, duration and severity. In some patients, diagnosis is made by Holter monitoring with the arrhythmia causing no symptoms while in other patients, syncope and, in severe cases, sudden cardiac death may result.10
Cerebrovascular diseases may be associated with a variety of alterations in respiratory patterns including CNH. It is a rare condition which is diagnosed by the following criteria: Hyperventilation that persists during sleep, low arterial paCO2, high arterial paO2 and alkalosis in the absence of drug administration or metabolic reasons.1,2 Alveolar hyperventilation decreases PaCO2 and increases the HCO3-/PaCO2 ratio, thus causing alkalosis. Significant alterations in extracellular pH produce reciprocal hydrogen and potassium ion shifts between the cells and the extracellular fluid. Alkalosis facilitates potassium movement into the cells and the resultant high intracellular concentration causes increased renal secretion leading to hypokalemia. Hypokalemia reduces the delayed rectifier potassium currents (IKr) and reduce repolarisation reserve. This prolongs cardiac repolarisation and QT interval which may precipitate TdP.11
Our patient presented with recurrent TdP with hemodynamic instability requiring cardioversion. Her restlessness and incoherent activity were attributed to cerebral hypoxia. Symptoms due to cerebral hypoxia of short duration are expected to improve promptly after restoration of stable hemodynamics. However in our patient neurological status did not improve even after achieving hemodynamically stable sinus rhythm for more than 24 hours. Hence we wanted to rule out any central nervous system lesion. A computed tomography scan of the brain revealed lower medullary ischemic infarct. Brain stem infarct may present with CNH which may mask the characteristic neurological deficits.1 The alkalosis and hypokalemia caused by CNH led to prolongation of QT interval and culminating in TdP. Incessant TdP episodes in our patient led to hemodynamic instability. The resultant cerebral hypoxia was implicated for her confusional state. Brain stem infarct, which was primarily responsible for TdP could be diagnosed only when confusional state persisted despite achieving stable hemodynamics. She ultimately succumbed to respiratory arrest after 48 hours of admission. The brain stem infarct presenting primarily as TdP is being reported for the first time.
|
References:
- Lee MC, Klassen AC and Resch JA. Respiratory Pattern Disturbances in Ischemic Cerebral Vascular Disease. Stroke 1974; 5: 612-616.
- Tarulli AW, Lim C, Bui JD, Saper CB and Alexander MP. Central Neurogenic Hyperventilation: A Case Report and Discussion of Pathophysiology. Arch Neurol 2005; 62: 1632-1634.
- Horacio JA and Nicolaos EM. Management Of Life Threatening Acid –Base Disorders: Second of Two Parts. N Engl J Med. 1998 May 28;338(22):1628-1629.
- Napolitano C, Priori SG, Schwartz PJ. Torsade de pointes. Mechanisms and management. Drugs. 1994 Jan;47(1):51-65.
-
Dessertenne F. La tachycardie ventriculaire a deux foyers opposes veriables. Arch Mal Coeur 1966; 59: 263–72
-
Jiashin W, Jianyi W, Douglas PZ. Early Afterdepolarizations, U Waves, and Torsades de Pointes. Circulation. 2002;105:675.
-
Dan MR and Mark EA. The pause that refreshes, or does it? Mechanisms in torsades de pointes. Heart 2000;84:235–237.
-
Roden DM, Lazzara R, Rosen MR, Schwartz PJ, Towbin J and Vincent GM for the SADS Foundation Task Force on LQTS. Multiple mechanisms in the long QT syndrome: current knowledge, gaps, and future directions. Circulation 1996;94:1996–2012.
-
Chiang CE, Roden DM. The Long QT Syndromes: Genetic Basis and Clinical Implications. J Am Coll Cardiol 2000;36:1–12.
|